
Hello, Sign in
My Account
My Account
New customer? Start here
Innovative Research with Long Synthetic Peptides
Over the past 20 years peptides and peptide-like compounds have been identified as a promising source for pharmaceutically interesting drug candidates. However, the manufacturing challenges that peptide chemists faced have impeded the wide spread development of peptide related therapeutics. Historically, the synthesis of longer peptides has been prohibited by synthetic inefficiencies associated with peptide aggregation, such as slow or incomplete coupling and/or deprotection reactions. However, new strategies have tempered the costs and resources required to achieve efficient and economically viable methods to prepare longer and ‘difficult peptides’, thereby permitting peptides to gain ground as an attractive area for drug development. This article reviews the latest advanced chemistries for manufacturing long peptides successfully.
Peptide Synthesis Schemes: t-Boc and Fmoc
In 1984, Bruce Merrifield, an American chemist from Rockefeller University (New York), won the Nobel Prize for his contribution to the advancement of peptide chemistry. He developed a solid phase peptide synthesis (SPPS) methodology for peptides, which uses a polymer with reactive sites (solid supports, insoluble resin supports) that chemically combine to form the developing peptide chain. Using Merrifield’s technique, the problems associated with low yields due to separation and purification are avoided. Because of the high insolubility of the polymer, it can be filtered and washed without mechanical losses.
SPPS consists of three distinct steps:
1) chain assembly on a resin;
2) simultaneous or sequential cleavage and deprotection of the fully protected chain bound to the resin; 3) purification and characterization of the target peptide. Various chemical strategies exist for the chain assembly and cleavage/deprotection steps, whereas the purification and characterization step is mainly based on high performance liquid chromatography (HPLC) and mass spectrometry (MS).
Two major chemistries for SPPS are Fmoc (base labile alpha-amino protecting group) and t-Boc (acid labile protecting group). Each method involves different resins and amino acid side-chain protection and consequent cleavage/deprotection steps. The t-Boc method uses a strong acid (hydrogen fluoride) containing anisole alone or anisole plus other scavengers, whereas peptide-resins assembled by Fmoc chemistry are cleaved by the milder Reagents K or R (a mixture of trifluoroacetic acid, thioanisole, EDT, phenol and water). Fmoc chemistry is known for generating peptides of higher quality and in greater yield than t-Boc chemistry. Impurities in t-Boc-synthesized peptides are mostly attributed to cleavage problems, dehydration and t-butylation. For peptide assembly, HBTU/HOBt, carbodiimide mediated coupling, and PyBOP/HOBt are the most popular elongation schemes. After cleavage from the resin, peptides are usually purified by reverse phase HPLC using columns such as C-18, C-8, and C-4.
Current Challenges and Improvements
SPPS is highly amenable to automation due to the repetitive nature of key steps and the reliability of the chemistry. These advantages give peptide chemists the opportunity to prepare a number of different kinds of peptides (i.e. cyclic, branched and long peptides). Efficiently extending the length of synthetic peptides manufactured by SPPS offers the tantalizing possibility of preparing novel therapeutic peptides as well as biologically relevant small proteins such as cytokines, hormones, and protein modification enzymes. However, synthesis of longer peptides (50-100 amino acid residues in length) has remained more challenging due to problematic or inefficient coupling reactions.
Difficult couplings (Boc chemistry) and difficult deprotection/couplings (Fmoc chemistry) generate either deleted or truncated polypeptides. These side products and several additional impurities produced mainly during the final acidolytic cleavage of peptide-resin (e.g. side chain alkylation of Trp, Tyr, Cys and Met, and impurities due to aspartimide formation) are present in the crude together with the target polypeptide. A stretch of amino acid residues in a polypeptide deprotection and/or coupling reactions that does not undergo completion, or proceeds in low yield, is said to constitute a 'difficult sequence'. These are usually characterized by a stretch of 2-5 residues and occur typically at 5-15 residues from the C-terminus of a sequence being synthesized. They originate from inter-molecular aggregation of the resin-bound polypeptides. The alpha-amino groups of the growing chains are partially buried so that amide bond formation occurs through reactions with the incoming activated amino acids is mire difficult. Because of these difficulties. It had been held that polypeptides with length > 40-50 residue are infeasible to synthesize in the yield and purity required for therapeutic applications. Optimized Boc nd Fmoc strategies have in part helped to overcome these challenges.
The optimized protocols developed over the years for both Boc and Fmoc strategies have resulted in a significant increase in peptide length achievability.
New chemical ligation strategies and commercially available pseudoproline dipeptides have considerably changed long peptide synthesis in recent years. Pseudoproline dipeptides were introduced by Mutter et al. as reversible proline mimetics for modulation of protein structure. The proline-like moiety is generated by formation of an oxazolidine ring between the alpha-amino and the side chain hydroxyl groups of Ser or Thr with an aldehyde or ketone (Figure 1).
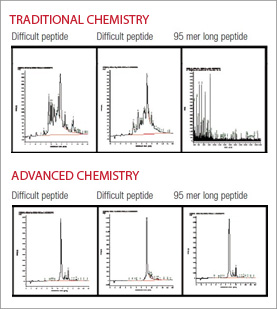
Such pseudoproline dipeptides are highly effective at preventing aggregation during solid phase synthesis as they disrupt the formation of beta-sheet and alpha-helical conformations. The effects can be long range, with the outset of aggregation often being postponed for as many as six residues, or eliminated altogether. They have been used with great effect to prepare long peptides/small proteins, cyclic peptides, or to disrupt structure as synthetic proline analogs. The incorporation of pseudoproline dipeptides has been found to lead to overall improvements in acylation and deprotection kinetics, resulting in better yield, purity and solubility of crude products and easier HPLC purification with higher amounts of isolated products (Figure 2).
Long peptides can also be obtained by chemical ligation. Ligation strategies have the considerable advantage that synthesis and purification of peptide segments up to 30 residues in length is straightforward but they are limited by the poor solubility of fully protected peptide segments and the tendency of alpha-car-boxy-activated peptides to racemize. These limitations have led a number of different research groups to develop methods based on the conjugation of unprotected peptide segments in aqueous media via formation of oxime, hydrazine or thiazolidine linkages, ulilizing the selectivity of the reaction of aldehydes with hydroxylamines, hydrazines and aminothiols in the presence of protonated amino functions, or by formation of an amide bond, through the intermediacy of a thioester or thiazolidine linkage. The original native chemical ligation (NCL) scheme for the preparation of small and medium size proteins is not restricted to a cysteine residue at the site of ligation anymore, offering subsequent flexibility for ligation site between polypeptides.
New Grounds for Peptide Research
Novel developments in peptide synthesis have opened new prospects in proteomics for studying post-translational modifications. For example, phosphorylated peptides are commonly used in kinase/phosphatase assays and antibody production. Furthermore, new caged phosphoserine peptides can be synthesized by synthetic methods in order to give researchers spatial and temporal control over release of the phosphorylated compound by UV irradiation. Construction of N-linked glycoproteins is performed using chemical ligation at an alanine or glycine site instead of the archetypal cysteine by selective desulfurization. Microwave-assisted solid phase peptide synthesis is also viable to produce such N-linked glycosylated polypeptides by reducing reaction time and improving yield and purity of final products. Chemoselective glycosylation, acylation, and alkylation of unprotected peptides can be accomplished by incorporation of N-alkylaminooxy amino acids into the peptide sequence to generate neoglyco- and neolipopetides. The ability to array such peptides allows for comprehensive studies of the effects of glycosylation and lipidation on peptide structure and function.
Tyrosine sulfation is a ubiquitous, reversible, post-translational modification found in many secreted and membrane-bound proteins. Post-assembly sulfation of peptides is always challenging because of the intrinsic acid lability of the O-sulfate linkage. Tyrosine sulfation can be protected by using a tyrosine sulfate ester moiety that is removed after deprotection, cleavage and purification of the sulfated peptide.
Many reagent and service companies have supplied academic and pharmaceutical researchers with modified peptides in order to develop quantitative assays for specific targets. The peptides used in these assays are often coupled to numerous dyes for monitoring biological activities. These peptides are also highly modified on amino side chains and sometimes the alpha-carbon backbone to mimic a substrate structure after modifications have been done. Such challenging reagents can now be made in large quantities at a reduced cost with the latest improvements in SPPS.
Conclusions
The rapid progress of SPPS for synthesizing polypeptides has opened new door in drug development for major pharmaceutical companies. Synthetic peptides as long as 40 residues are commercially available. Polypeptides over 100 residues are now a reality for large scale production, which can turn into more peptide candidates investigated in Phase II and III clinical trials. Chemokines, hormones and vaccines previously prepared through recombinant technologies are now revisited with peptide synthesis procedures.