
Hello, Sign in
My Account
My Account
New customer? Start here
Intrafamilial and interfamilial Bcl-2 protein contacts regulate key aspects of apoptosis. Corruption of apoptotic instructions is associated with a large subset of human diseases, ranging from cancer and cardiovascular to neurodegenerative diseases, and many others. Understanding regulation of apoptosis is critical to a strategy for pharmaceutical intervention. The BH3 domain of Bcl-2 family members is key to Bcl-2 regulatory function.
Apoptosis is a critical aspect of cellular function and human disease
Apoptosis (programmed cell death) is characterized by cell shrinkage, membrane blebbing, phagocytic engulfment of the fragmented cell, DNA fragmentation and mitochondrial release of cytochrome C. Tightly regulated termination is essential to cull unneeded, aging, mutated, or infected cells. Dysregulation of death/survival signals is implicated in a broad range of human diseases. Unchecked proliferation of cell growth is a key feature of cancer, autoimmune diseases, and viral infections. Tellingly, cancerous cells subjected to traditional cytotoxic therapies may develop resistance by evolving methods to subvert apoptosis. Conversely, apoptosis in irreplaceable cells or excessive apoptosis is implicated in neurodegenerative and cardiovascular disorders. Controlling fidelity of the apoptotic pathways may lead to new methods to treat challenging diseases.
Bcl-2 family proteins play pivotal roles in apoptosis
Founding family member Bcl-2 is overexpressed in 50% of all cancers, including ~70% of breast cancers, ~30%-60% of prostate cancers, ~90% of colorectal cancers, ~60% of gastric cancers, ~100% of small-cell lung carcinomas, ~20% of non-small-cell lung cancers, ~30% of neuroblastomas, and ~80% of B cell lymphomas. Bcl-2’s ability to impair apoptosis induction by traditional cytotoxic (chemotherapeutic) drugs is well-established. Tumor cells gain resistance to therapy by reducing expression of pro-apoptotic Bcl-2 protein family members like Bax. Bcl-2 antisense olignonucleotides inhibit non-Hodgkin’s lymphoma in humans and enhance tumor cell susceptibility to chemotherapeutics.
Pro-apoptotic members, including Bax, Bak, Bid, and Bim, trigger release of death-including proteins from mitochondria, while anti-apoptotic members such as Bcl-2 and Bcl-xL inhibit release. These death-inducing proteins work through pathways including caspase activation and DNA fragmentation.
Bcl-2 family proteins interact through a network of homo- and heterodimers that have been observed in yeast two-hybrid assays, coimmunoprecipitation from mammalian cells, and intact mitochondria in vivo. Mutagenesis studies suggest homo- and heterodimerization events are critical to function. A role for homo- and hetero-dismerization in mitochondrial pore formation that is necessary for cytochrome C efflux has been postulated.
BH3 domain interaction is the key regulatory element in Bcl-2 family member proteins
There are four conserved homologous motifs within the Bcl-2 family: BH1, BH2, BH3, and BH4. The BH3 domain is critical for Bcl-2 family heterodimerization and death-promoting activity. Bid, Bcl-2, and Bcl-xL cleavage exposes the BH3 domain and recruits these molecules to mediate apoptosis. Some Bcl-2 family members, including Bik, Bid, and Hrk, contain only the BH3 domain. Deletion of BH3 domains from this subfamily abolishes both ability to promote cell death and heterodimerization with anti-apoptotic proteins. Overexpression of Bak Bh3 domain fragments induces mammalian cell death.
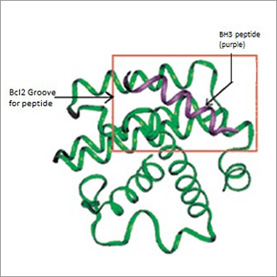
The Bcl-xL structure reveals a receptor-like hydrophobic groove formed by the combination of the BH1, BH2, and BH3 domains, binding epitopes located on dimerizing partner proteins. The BH3 domain inserts into the surface pocket on Bcl-xL, similar to a peptide ligand. Death agonists such as Bax, Bak and Bad, insert via BH3 domains into the groove of Bcl-2 or Bcl-xL and promote apoptosis. A Bcl-xL:Bak complex structure confirms the critical nature of BH3 contacts.
Study of BH3 domain-based interactions will permit elucidation of multiple protein-protein interactions that delineate key apoptotic pathways
Functional and structural evidence suggests that BH3 domains are pivotal to Bcl-2 regulated apoptosis. BH3 peptides that bind the Bcl-2 pocket block functional protein-protein interactions, implying that secondary and tertiary domain structure are retained in peptidic versions. BH3 subunits of Bak, Bax, or Bid induce apoptosis by causing rapid activation of caspases, whereas a Bak BH3 mutant peptide containing an Ala substitution al Leu-78, which is critical for Bcl-xL binding, does not show any effect. Bak, Bax, and Bad BH3 peptides block the heterodimerization of Bcl-xL with cell death agonists in a dose-dependent manner in an in vitro assay. Bad BH3 peptides are more potent than other Bcl-2 family BH3 domains in blocking protein-protein interactions of Bcl-xL. Bad and Bax BH3 peptides block Bcl-2:Bak association and induce apoptosis in prostate carcinoma cells, which is clocked by caspase inhibitors.
BH3 peptide structure has been characterized by different biophysical methods. The high affinity of a Bak BH3 peptide for Bcl-xL was explained by the NMR structure of a Bcl-xL:bak BH3 peptide complex. A murine Bad Bh3 peptide was also determined by NMR spectroscopy and was similar to the structure of the peptide in a Bak BH3 peptide:Bcl-xL complex. A crystal structure of Bcl-xL in complex with a peptide derived from the bH3 domain of Bak has been solved. The structure reveals a hydrophobic surface pocket on Bcl-xL formed by the BH1-3 domains bound by the Bak BH3 domain peptide in helical conformation.
Inhibitors of Bcl-2 protein-protein interactions may provide useful leads for drug design
Nonpeptidic small molecules that target BH3 binding are valuable as probes for mapping Bcl-2 family protein binding pockets and as leads for new therapeutic agents. Abnormal Bcl-2 gene expression is found in ~50% of all cancers. Bcl-2 protein levels correlate with resistance to chemotherapeutic and radiation therapies. Bcl-2 protein inhibitors may be more selective than conventional cytotoxic chemotherapies, since Bcl-2 is low in most normal cell types. Antisense oligonucleotides targeted against the Bcl-2 gene specifically inhibit non-Hodgkin’s lymphoma in humans, validating Bcl-2 as a therapeutic target. Pro-apoptotic proteins such as Bax and Bad are attractive targets for diseases where the goal is to prevent excessive cell death, such as cardiovascular and neurodegenerative diseases.
The first reported inhibitor of Bcl-2 (HA14-1) was discovered by computer screening. It induces apoptosis (human myeoloid leukemia, breast cancer, prostate cancer), and shows synergistic activity with an MEK inhibitor in diminishing the CFU-blast of primary AML patients. Since then, dozens of nonpeptidic Bcl-2 inhibitors have been identified using a variety of techniques, including direct ligand binding measurements based on fluorescence emission, NMR chemical shift titration, competitive binding assays, and computer modeling approaches. These inhibitors interact with Bcl-2 and Bcl-xL, and inhibit apoptosis in model systems.